tfcb_2020
Lecture 13: In-class exercises
PUT YOUR NAME HERE 11/12/2020
Load packages you need
library(tidyverse)
## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.0 ──
## ✓ ggplot2 3.3.2 ✓ purrr 0.3.4
## ✓ tibble 3.0.3 ✓ dplyr 1.0.2
## ✓ tidyr 1.1.2 ✓ stringr 1.4.0
## ✓ readr 1.3.1 ✓ forcats 0.5.0
## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## x dplyr::filter() masks stats::filter()
## x dplyr::lag() masks stats::lag()
Read in example dataset 5 in the data subfolder and store the result in flow_data variable
Note that the first line is a comment that mentions the column names.
This annoying structure is typical of datasets that you might receive
from someone else. Look at the documentation of read_csv and figure
out how to skip commented out lines and assign your own column
names.
flow_data <- read_csv("data/example_dataset_5.csv", col_names = c("strain", "yfp", "rfp", "replicate"), comment = "#") %>%
print()
## Parsed with column specification:
## cols(
## strain = col_character(),
## yfp = col_double(),
## rfp = col_double(),
## replicate = col_double()
## )
## # A tibble: 74 x 4
## strain yfp rfp replicate
## <chr> <dbl> <dbl> <dbl>
## 1 schp677 4123 20661 1
## 2 schp678 4550 21437 1
## 3 schp675 3880 21323 1
## 4 schp676 2863 20668 1
## 5 schp687 4767 20995 1
## 6 schp688 1274 20927 1
## 7 schp679 2605 20840 1
## 8 schp680 1175 20902 1
## 9 schp681 3861 20659 1
## 10 schp683 9949 25406 1
## # … with 64 more rows
Read in example dataset 3 in the data subfolder which contains the annotations for the above table and store it in a variable annotations
annotations <- read_tsv("data/example_dataset_3.tsv") %>%
print()
## Parsed with column specification:
## cols(
## strain = col_character(),
## insert_sequence = col_character(),
## kozak_region = col_character()
## )
## # A tibble: 17 x 3
## strain insert_sequence kozak_region
## <chr> <chr> <chr>
## 1 schp674 10×AAG G
## 2 schp675 10×AAG B
## 3 schp676 10×AAG F
## 4 schp677 10×AAG E
## 5 schp678 10×AAG D
## 6 schp679 10×AAG A
## 7 schp680 10×AAG H
## 8 schp681 10×AAG C
## 9 schp683 10×AGA G
## 10 schp684 10×AGA B
## 11 schp685 10×AGA F
## 12 schp686 10×AGA E
## 13 schp687 10×AGA D
## 14 schp688 10×AGA A
## 15 schp689 10×AGA H
## 16 schp690 10×AGA C
## 17 control <NA> <NA>
Join the two tables above and assign to a new variable data
data <- inner_join(flow_data, annotations, by = "strain") %>%
print()
## # A tibble: 74 x 6
## strain yfp rfp replicate insert_sequence kozak_region
## <chr> <dbl> <dbl> <dbl> <chr> <chr>
## 1 schp677 4123 20661 1 10×AAG E
## 2 schp678 4550 21437 1 10×AAG D
## 3 schp675 3880 21323 1 10×AAG B
## 4 schp676 2863 20668 1 10×AAG F
## 5 schp687 4767 20995 1 10×AGA D
## 6 schp688 1274 20927 1 10×AGA A
## 7 schp679 2605 20840 1 10×AAG A
## 8 schp680 1175 20902 1 10×AAG H
## 9 schp681 3861 20659 1 10×AAG C
## 10 schp683 9949 25406 1 10×AGA G
## # … with 64 more rows
Create a new column ratio containing ratio of YFP and RFP signals for each strain replicate.
Store the result in the same data variable.
data <- data %>%
mutate(ratio = yfp / rfp) %>%
print()
## # A tibble: 74 x 7
## strain yfp rfp replicate insert_sequence kozak_region ratio
## <chr> <dbl> <dbl> <dbl> <chr> <chr> <dbl>
## 1 schp677 4123 20661 1 10×AAG E 0.200
## 2 schp678 4550 21437 1 10×AAG D 0.212
## 3 schp675 3880 21323 1 10×AAG B 0.182
## 4 schp676 2863 20668 1 10×AAG F 0.139
## 5 schp687 4767 20995 1 10×AGA D 0.227
## 6 schp688 1274 20927 1 10×AGA A 0.0609
## 7 schp679 2605 20840 1 10×AAG A 0.125
## 8 schp680 1175 20902 1 10×AAG H 0.0562
## 9 schp681 3861 20659 1 10×AAG C 0.187
## 10 schp683 9949 25406 1 10×AGA G 0.392
## # … with 64 more rows
Calculate the mean and standard deviation of the YFP-RFP ratio across all replicates for each strain.
To do the above, create new summary columns mean_ratio and std_ratio
after grouping all replicates.
Assign the result to avg_data variable.
avg_data <- data %>%
group_by(strain) %>%
summarize(mean_ratio = mean(ratio), std_ratio = sd(ratio)) %>%
print()
## `summarise()` ungrouping output (override with `.groups` argument)
## # A tibble: 16 x 3
## strain mean_ratio std_ratio
## <chr> <dbl> <dbl>
## 1 schp674 0.0625 0.000640
## 2 schp675 0.181 0.00887
## 3 schp676 0.131 0.0118
## 4 schp677 0.193 0.00747
## 5 schp678 0.212 0.00223
## 6 schp679 0.128 0.0125
## 7 schp680 0.0578 0.00256
## 8 schp681 0.183 0.00520
## 9 schp683 0.392 0.0246
## 10 schp684 0.160 0.00517
## 11 schp685 0.322 0.0124
## 12 schp686 0.236 0.00584
## 13 schp687 0.223 0.00523
## 14 schp688 0.0841 0.0163
## 15 schp689 0.381 0.0311
## 16 schp690 0.172 0.00442
What happened to the annotations? Can you join them back with avg_data?
avg_data <- inner_join(avg_data, annotations, by = "strain") %>%
print()
## # A tibble: 16 x 5
## strain mean_ratio std_ratio insert_sequence kozak_region
## <chr> <dbl> <dbl> <chr> <chr>
## 1 schp674 0.0625 0.000640 10×AAG G
## 2 schp675 0.181 0.00887 10×AAG B
## 3 schp676 0.131 0.0118 10×AAG F
## 4 schp677 0.193 0.00747 10×AAG E
## 5 schp678 0.212 0.00223 10×AAG D
## 6 schp679 0.128 0.0125 10×AAG A
## 7 schp680 0.0578 0.00256 10×AAG H
## 8 schp681 0.183 0.00520 10×AAG C
## 9 schp683 0.392 0.0246 10×AGA G
## 10 schp684 0.160 0.00517 10×AGA B
## 11 schp685 0.322 0.0124 10×AGA F
## 12 schp686 0.236 0.00584 10×AGA E
## 13 schp687 0.223 0.00523 10×AGA D
## 14 schp688 0.0841 0.0163 10×AGA A
## 15 schp689 0.381 0.0311 10×AGA H
## 16 schp690 0.172 0.00442 10×AGA C
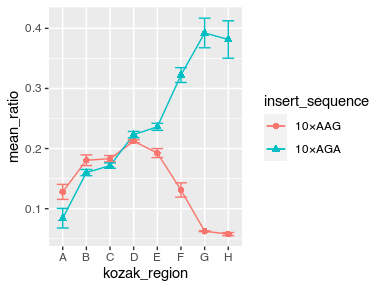
Plot the mean and standard deviation of the YFP-RFP ratio as a function of the Kozak region.
Display the result as a point graph and a line graph with error bars around the mean.
Give the insert_sequences different shapes and colors.
Can you make the markers twice their default size?
Can you make the error bars half as wide as their default width?
Store the result as a PDF file named demo_plot.pdf in figures
subfolder.
avg_data %>%
ggplot(aes(x = kozak_region, y = mean_ratio, color = insert_sequence, shape = insert_sequence, group = insert_sequence)) +
geom_point(size = 2) +
geom_line() +
geom_errorbar(aes(ymin = mean_ratio - std_ratio, ymax = mean_ratio + std_ratio), width = 0.5)
ggsave("figures/example_plot.pdf")
## Saving 4 x 3 in image