Classifying cells with Garnett
So far we have:
- imported a CDS object
- corrected for batch effects
- put our data into lower dimensional space
- clustered and visualized our cells in UMAP space
Today we will learn how to:
- use Garnett to classify cells with a pre-trained classifier and by training our own classifier
- visualize cells with cell labels
We can run clustering algorithms on our cells to identify “cell types” or cells that are similar to each other based on their shared gene expression. Without manually inspecting what genes are unique to cluster, it is difficult to assign cell types. Manually assigning cell clusters to known cell types based on canonical gene expression is a very laborious process. Luckily, the Trapnell lab has developed the software package Garnett.
Citation:
Pliner, H.A., Shendure, J. & Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat Methods 16, 983–986 (2019). https://doi.org/10.1038/s41592-019-0535-3
Garnett is a software package that facilitates automated cell type classification from single cell gene expression data. Garnett works by taking single-cell data, along with a cell type definition (marker) file, and training a regression-based classifier. Once a classifier is trained for a tissue/sample type, it can be applied to classify future datasets from similar tissues. In addition to describing training and classifying functions, this website aims to be a repository of previously trained classifiers.
There are two options when you are starting:
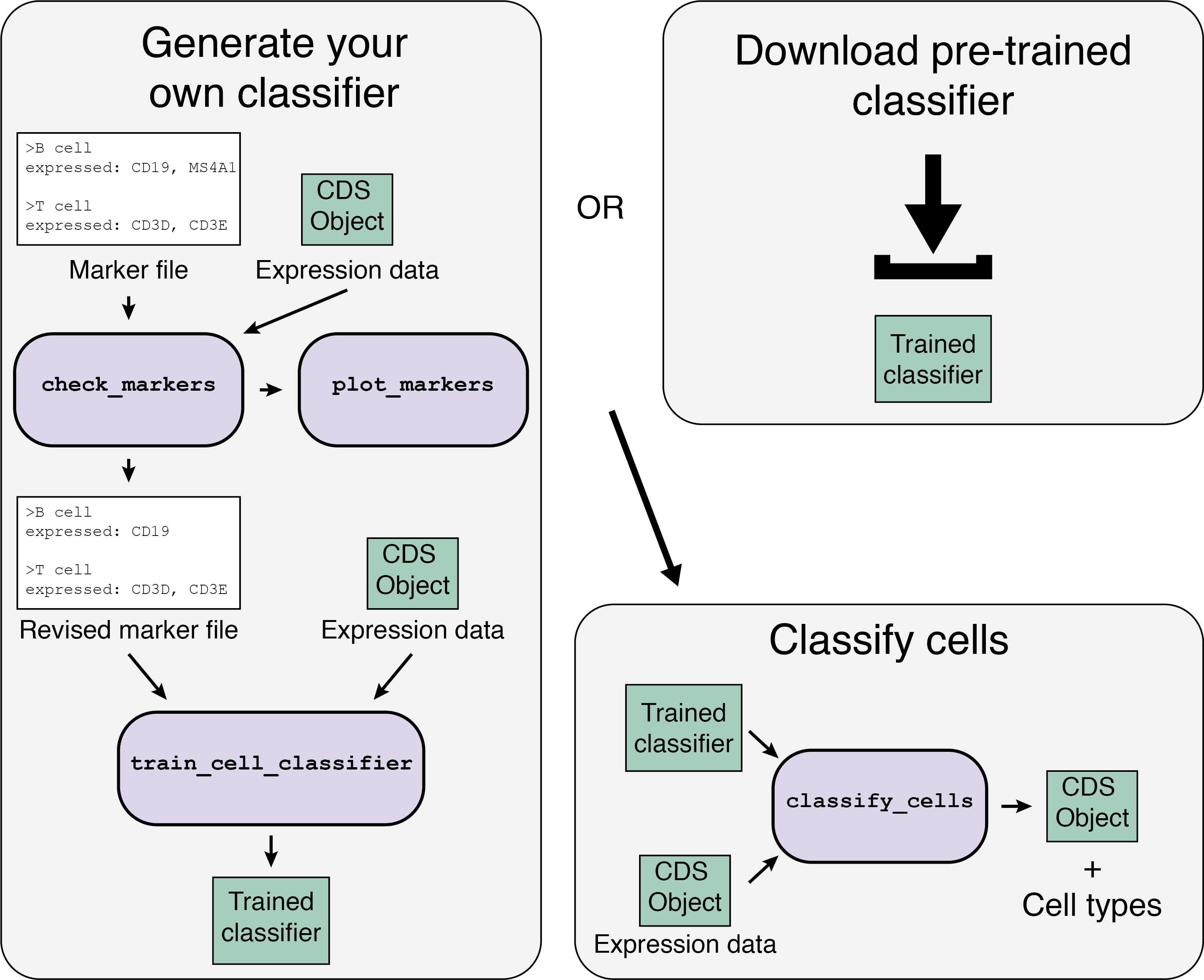 Image Source: https://cole-trapnell-lab.github.io/garnett/classifiers/
Image Source: https://cole-trapnell-lab.github.io/garnett/classifiers/
# ignore warnings
Sys.setenv(R_REMOTES_NO_ERRORS_FROM_WARNINGS="true")
# load the necessary packages
library(monocle3)
# install garnett
devtools::install_github("cole-trapnell-lab/garnett", ref="monocle3")
library(garnett)
# install Mouse gene annotation
BiocManager::install(c('org.Mm.eg.db'))
library("org.Mm.eg.db")
BiocManager::install(c('ggplot2'))
library(ggplot2)
# use function install.packages("package_name") if package not previously installed
library(tidyverse)
library(ggplot2)
library(tidyr)
library(viridis)
library(ggridges)
library(RColorBrewer)
library(ggrepel)
library(pheatmap)
The Garnett workflow has two major parts, each described in detail below:
Train/obtain the classifier:
- Download an existing classifier, or train your own. To train, Garnett parses a marker file, chooses a set of training cells, and then trains a multinomial classifier to distinguish cell types.
- Classify cells: Next, Garnett applies the classifier to a set of cells to generate cell type assignments. Optionally, Garnett will expand classifications to similar cells to generate a separate set of cluster-extended type assignments.
First, let’s download the “mmlung” marker file (https://cole-trapnell-lab.github.io/garnett/classifiers/). Save this marker file to your “scRNAseq_project” folder. Open up the marker file and take a look at it.
The marker file contains a list of cell type definitions written in an easy-to-read text format. The cell type definitions tell Garnett how to choose cells to train the model on. Each cell type definition starts with a ‘>’ symbol and the cell type name, followed by a series of lines with definition information. Definition lines start with a keyword and a ‘:’ and entries are separated by a comma. Note: the Garnett syntax allows for entries following the ‘:’ to move onto following lines however, you may not move to a new line mid entry (i.e. you can go to a new line only after a comma).
There are several ways to define cell types in the Garnett marker file format. In general, each cell’s definition can have three major components. Only the first component is required. The first and most important specification for a cell type is its expression. Garnett offers several options for specifying marker genes, detailed below.
Format:
expressed: gene1, gene2
not expressed: gene1, gene2
Example:
expressed: MYOD1, MYH3
not expressed: PAX6, PAX3
In addition to expression information, you can further refine your cell type definitions using meta data. This is also where you will specify any subtypes you expect in your data.
subtype of: allows you to specify that a cell type is a subtype of another cell type in your definition file.
custom meta data: specification allows you to provide any further meta data requirements for your cell type. Any column in the pData table of your CDS object can be used as a meta data specification. In the example above, there would be a column in the pData table called “tissue”.
Lastly, we highly recommend that you document how you chose your marker definitions. To make it easier to keep track of, we provide an additional specification - references: - that will store your citation information for each cell type.
A more complex example:
B cells
expressed: CD19, MS4A1
expressed above: CD79A 10
references: https://www.abcam.com/primary-antibodies/b-cells-basic-immunophenotyping, 10.3109/07420528.2013.775654
T cells
expressed: CD3D
sample: blood # A meta data specification
Helper T cells
expressed: CD4
subtype of: T cells
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=12000723
Classify our cells by training our own classifier with a marker file
# import cds we saved at the end of lesson 2
cds <- readRDS(<filepath>/<filename>.rds)
# define marker file path
marker_file_path <- "/Users/elizabarkan/Desktop/mmLung_markers.txt"
# check our markers
marker_check <- check_markers(cds, marker_file_path,
db=org.Mm.eg.db,
cds_gene_id_type = "SYMBOL", # in cds: read gene symbol or ensembl id
marker_file_gene_id_type = "SYMBOL") # in marker file: read gene symbol or ensembl id
# check ambiguity scores for markers (lower ambiguity scores are more desirable
plot_markers(marker_check)
(insert something about how to deal with high ambiguity scores)
- maybe make some corrections to the marker file to make it better
```{r}
train a classifier with our marker file
our_lung_classifier <- train_cell_classifier(cds = cds, marker_file = marker_file_path, db=org.Mm.eg.db,, cds_gene_id_type = “SYMBOL”, num_unknown = 50, marker_file_gene_id_type = “SYMBOL”)
run the classifier on the cells in our cds
cds_manual <- classify_cells(cds, our_lung_classifier, db=org.Mm.eg.db, cluster_extend = TRUE, cds_gene_id_type = “SYMBOL”)
look at cell classifications
head(pData(cds_manual))
table of how many cells are classified into each major type
table(pData(cds_manual)$cell_type)
table of how many cells are classified into each sub type
table(pData(cds_manual)$cluster_ext_type)
Next, let's visualize cells colored by cell type
```{r}
# visualize cells by major cell type
plot_cells(cds_manual, color_cells_by = "cell_type") + theme_bw()

# visualize cells by cell sub type
plot_cells(cds_manual, color_cells_by = "cell_type") + theme_bw()

Classify cells with a pre-trained classifier
Alternatively, we could have used the classifier already trained on cells, instead of training our own classifier. Luckily, in our case there exists a classifier run on cells of the same species, age and tissue but this is not always the case. Submit your marker file and classifiers if you make a new one to help increase the size of the marker file and classifier database (https://cole-trapnell-lab.github.io/garnett/docs/#submitting-a-classifier)!
Let’s download the “mmlung” classifier (https://cole-trapnell-lab.github.io/garnett/classifiers/). Save this classifier to your “scRNAseq_project” folder.
lung_classifier <- readRDS("<filepath>/mmLung_classifier.RDS")
Let’s classify the cells with the lung classifier we imported
cds_auto <- classify_cells(cds, lung_classifier,
db=org.Mm.eg.db,
cluster_extend = TRUE,
cds_gene_id_type = "SYMBOL")
head(pData(cds_auto))
table(pData(cds_auto)$cell_type)
table(pData(cds_auto)$cluster_ext_type)
Next, let’s visualize cells colored by cell type
plot_cells(cds_auto, color_cells_by = "cell_type") + theme_bw()

plot_cells(cds_auto, color_cells_by = "cell_type") + theme_bw()

Visualize Different Annotations of the Same Cells with Different Annotation Methods
Say we have two methods of annotating cells and we want to compare them– specifically identify which cell types are commonly labaled as one cell type with one method and another cell type with another method. For example, it may be that both classifiers do a great job with classifying one cell type but another cell type is more ambigous so the classifier perform differently. We can create a “Confusion Matrix” to visualize these differences.
Code provided by Sanjay Srivatsan
# append cell annotations to the original cds
cds$manual_cell_type = pData(cds_manual)$cell_type
cds$manual_cluster_ext_type = pData(cds_manual)$cluster_ext_type
cds$auto_cell_type = pData(cds_auto)$cell_type
cds$auto_cluster_ext_type = pData(cds_auto)$cluster_ext_type
# Make a matrix to compare manual vs. already made classifier (auto)
matrix_for_heatmap =
colData(cds) %>%
as.data.frame() %>%
group_by(manual_cluster_ext_type) %>%
add_tally(name = "num_manual_cell_type") %>%
ungroup() %>%
group_by(manual_cluster_ext_type, auto_cluster_ext_type) %>%
mutate(percent_nn_in_garnett = n()/num_manual_cell_type) %>%
dplyr::select(auto_cluster_ext_type, manual_cluster_ext_type, percent_nn_in_garnett) %>%
distinct() %>%
spread(key =auto_cluster_ext_type, value = percent_nn_in_garnett, fill = 0)
# create matrix for heatmap
matrix_for_heatmap =
matrix_for_heatmap %>%
tibble::column_to_rownames(var = "manual_cluster_ext_type") %>%
as.matrix()
# save heatmap as pdf
pheatmap(matrix_for_heatmap,
cellwidth = 15,
cellheight = 15,
cluster_rows = F,
cluster_cols = F,
fontsize_row = 10,
fontsize_col = 10,
legend = T,
filename = "<filepath</<filename>.pdf",
color = viridis(option = "viridis", n = 40))

Check that the heatmap PDF saved in your specified file path and that it looks like the heatmap above. Examine the heatmap and take a few minutes and make a note of what you observe – specifically what cell types are classified
Save CDS
We will be using this CDS with cell type labels for the next class
saveRDS(cds, "<filepath>/cds_class3.RDS")
Wrapping Up
Today we learned how to:
- use Garnett to classify cells with a pre-trained classifier and by training our own classifier
- visualize cells with cell labels