Making Cell Data Set objects for Single Cell Data Analysis in Monocle
Two weeks before session, distribute instructions on how to install Monocle version 3 (https://cole-trapnell-lab.github.io/monocle3/docs/installation/), common troubleshooting (https://cole-trapnell-lab.github.io/monocle3/docs/installation/#troubleshooting) and answers to common questions and issues (https://github.com/cole-trapnell-lab/monocle3/issues).
One week before session, hold an office hour to answer any remaining questions regarding Monocle installation. Students are ready to proceed with the course once they can run library(monocle3) without any errors. __________________________
<FIXME: add introduction to scRNAseq and why this data is valuable>
Today we will learn how to:
- create the data structures (CDS) that contain our single cell RNA-sequencing data starting with different data formats
- subset a CDS on a particular column value
- merge multiple CDSs
This class is based on materials provided by Monocle 3 and the Brotman Baty Institute (Aishwarya Gogate & Hannah Pliner).
Cell data sets or CDSs are data structures that contain all of our single cell RNA-sequencing data. These CDS objects are multi-dimensional and are made up of 3 individual files:
- a matrix of counts of sequenced reads or unique molecular identifiers (UMIs) by genes (rows) and cell (columns)
- a list of gene IDs that represents all genes quantified (correspond to matrix rows)
- a list of cell IDs that represents all cells measured (correspond to matrix columns) and any additional single cell data (i.e. replicates, experimental conditions) associated with those cell IDs
We can start by checking that Monocle has been installed correctly. Start a new R Markdown document so you can code along. If the command library(monocle3) runs without any errors, Monocle 3 has been successfully installed!
library(monocle3)
Monocle is a toolkit in R to analyze single-cell gene expression experiments.
Citations:
Trapnell, C., Cacchiarelli, D., Grimsby, J. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). https://doi.org/10.1038/nbt.2859
Qiu, X., Hill, A., Packer, J. et al. Single-cell mRNA quantification and differential analysis with Census. Nat Methods 14, 309–315 (2017). https://doi.org/10.1038/nmeth.4150
Qiu, X., Mao, Q., Tang, Y. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982 (2017). https://doi.org/10.1038/nmeth.4402
Cao, J., Spielmann, M., Qiu, X. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). https://doi.org/10.1038/s41586-019-0969-x
To get started, we want to download the single cell gene expression data we will be working with today. This data is from mouse and has been processed with 10x Genomics (v2) single cell platform.
- Download the data as a zip from Github (https://github.com/fredhutchio/scRNAseq/tree/monocle/lung_data/Lung-10X_P7_8)
- Unzip the data
- Move the data to a new folder titled “scRNAseq_class”
There are multiple methods to create a CDS object in Monocle that we will cover today- if you are starting with:
- full matrix of counts + cell IDs + gene IDs
- sparse matrix of counts + cell IDs + gene IDs
- 10X Genomics CellRanger output a. file path to the ‘outs’ file b. individual CellRanger output components
- CDS object
1. Starting with a full matrix of counts + cell IDs + gene IDs
First we will assume you are starting with the least processed form of single cell gene expression data, which is a matrix of counts- either reads or UMIs. To load the data into a CDS, you must convert the matrix into a sparse matrix. Instead of a matrix of values, a sparse matrix has three columns: row, column and value. The position of each non-zero value in the matrix is stored in this format. If you have a lot of data and a lot of that data is made of zeros, this is a much more efficient way to store the data AND it is the input format neccessary to make a CDS object for Monocle.
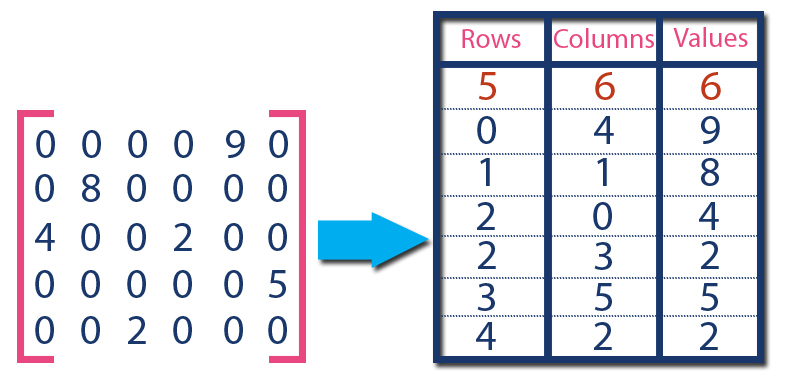 Image Source: www.btechsmartclass.com
Image Source: www.btechsmartclass.com
# load packages
library(Matrix)
Import the data into R markdown.
cell_metadata = read.table("<filepath>/barcodes.tsv", sep = "\t", header = FALSE)
gene_annotation = read.table("<filepath>/genes.tsv", sep = "\t", header = FALSE)
expression_matrix = read.table("<filepath>/dense_matrix.txt", sep = " ", header = FALSE)
# inspect the dense matrix format
print(expression_matrix)
Reformat the data around the following priniples:
- Gene names must have a column name “gene_short_name”
gene_annotation = rename(gene_annotation, c("V1"="gene_short_name")) - expression matrix column names must match the row names of cell metadata
colnames(expression_matrix) <- seq(1, dim(cell_metadata)[1], by=1) row.names(cell_metadata) = seq(1, dim(cell_metadata)[1], by=1) - Convert the regular matrix to a sparse matrix
data <- as(as.matrix(expression_matrix), 'sparseMatrix') - For Monocle version 3, cell metadata and gene annotation need to be converted to a data frame
pd <- data.frame(cell_metadata) fData <- data.frame(gene_short_name = gene_annotation$gene_short_name, row.names = row.names(data))Now we can make the CDS object. Once we have a CDS object, we can
cds <- new_cell_data_set(data, cell_metadata = pd, gene_metadata = fData) cds # look at the cds_object and confirm we have correct number of rows and columnsNow we just made a CDS object the hard waty but there are a few other ways to make a CDS object. You might be starting with a sparse matrix already
2. Starting with sparse matrix of counts + cell IDs + gene IDs
If we are starting with a matrix that is already sparse, we only need to rerun steps 1, 2 and 4 (skip 3). We also will use the readMM function from the Matrix package to read a sparse matrix file into R. ```{r} cell_metadata = read.table(“
/5968960/droplet/Lung-10X_P7_8/barcodes.tsv", sep = "\t", header = FALSE) gene_annotation = read.table(" /5968960/droplet/Lung-10X_P7_8/genes.tsv", sep = "\t", header = FALSE) sparse_matrix = readMM(" /5968960/droplet/Lung-10X_P7_8/matrix.mtx") # different from 1, .mtx file type is a sparseMatrix format
inspect the sparse matrix format
print(sparse_matrix)
1 - Gene names must have a column name “gene_short_name”
gene_annotation = rename(gene_annotation, c(“V1”=”gene_short_name”))
2 - expression matrix column names must match the row names of cell metadata
colnames(sparse_matrix) <- seq(1, dim(cell_metadata)[1], by=1) # different from 1 row.names(cell_metadata) = seq(1, dim(cell_metadata)[1], by=1)
4 - for Monocle version 3, cell metadata and gene annotation need to be converted to a data frame
pd <- data.frame(cell_metadata) fData <- data.frame(gene_short_name = gene_annotation$gene_short_name, row.names = row.names(sparse_matrix))
make CDS object
cds <- new_cell_data_set(sparse_matrix, cell_metadata = pd, gene_metadata = fData) cds # look at the cds_object and confirm we have correct number of rows and columns
## 3. Starting with 10X Genomics CellRanger output
A lot of researchers work with data from a CellRanger output from 10x so now we will introduce how to import data from a cell ranger output. There are two ways to do this, the first is to provide Monocle with the path to the 'outs' directory created by cell ranger. It is import that your directory structure match the directory and file names in order for this function to work. Take a few minutes to create this data structure and copy and paste your three files in the final directory.
With 10x v2 data:
- 10x_data/outs/filtered_gene_bc_matrices/"genome"/barcodes.tsv
- 10x_data/outs/filtered_gene_bc_matrices/"genome"/genes.tsv
- 10x_data/outs/filtered_gene_bc_matrices/"genome"/matrix.mtx
With 10x v3 data:
- 10x_data/outs/filtered_feature_bc_matrices/barcodes.tsv.gz
- 10x_data/outs/filtered_feature_bc_matrices/features.tsv.gz
- 10x_data/outs/filtered_feature_bc_matrices/matrix.mtx.gz
```{r}
# our data is 10x v2 data
cds <- load_cellranger_data(pipestance_path ="<filepath>/5968960/droplet/Lung-10X_P7_8/10x_data")
cds # look at the cds_object and confirm we have correct number of rows and columns
Alternatively, we can provide Monocle with the individual file paths for each of the three components.
cds <- load_mm_data(mat_path = "<filepath>/10x_data/outs/filtered_gene_bc_matrices/"genome"/matrix.mtx",feature_anno_path = "<filepath>/10x_data/outs/filtered_gene_bc_matrices/"genome"/genes.tsv", cell_anno_path = "<filepath>/10x_data/outs/filtered_gene_bc_matrices/"genome"/barcodes.tsv")
names(rowData(cds_obj))[names(rowData(cds_obj))=="V2"] <- "gene_short_name"
cds # look at the cds_object and confirm we have correct number of rows and columns
4. Starting with CDS object
Lastly, if someone has already saved a CDS object, you can import it directly into R.
cds <- readRDS("<filepath>/<filename>.RDS")
names(rowData(cds_obj))[names(rowData(cds_obj))=="V2"] <- "gene_short_name"
cds # look at the cds_object and confirm we have correct number of rows and columns
The CDS object
Now that we have a CDS object, let’s get familiar with it!
cds
We can see the object is made up of the following multidimensions components:
- class: cell_data_set
- dimensions: n rows (genes) and m columns (cells)
- metadata: version of CDS object
- assays: the types of measurements made for the cells, in this case we just have UMI counts
- rownames: gene names
- rowData names: column names from gene input
- colnames: cell ID names, in this case they are the actual unique cell barcodes
- colData names: column names from cell metadata input,
- reducedDimNames: once you run dimension reductions (e.g. PCA, tSNE, UMAP), the values for those reduced dimensions will be stored here
- spikeNames: ? (fixme)
- altExpNames: ? (fixme)
Save CDS for next class
We will be using this CDS object in the following classes
saveRDS(cds,"<filepath>/<filename>.RDS")
Subsetting and Merging CDS objects
In the next couple classes we are going to show you how to use Monocle to clean up, visualize and run statistical tests on your data. You may find you need to subset your CDS object to process different types of data individually or you may want to combine CDS objects.
fix me: make these examples more interesting, relevant
# example of subsetting a cds to remove cells without a lot of UMIs, size factors are used to adjust for UMI count
cds_high_sf <- cds_obj[colData(cds_obj)$n.umi > 1000,]
# subset CDS by cells into a new CDS object
cds2 <- cds[,1:100] # cds2 contains the first 100 cells
# merge two CDS objects
big_cds <- combine_cds(list(cds, cds2)) # combine them into one CDS
If you want to learn more about a function in R you can put a question mark in front of to learn what arguments you can pass to it. For example with combine_cds, you can optionally specify ‘cell_names_unique’ to specify if Monocle should assume your Cell IDs are from the same cells (TRUE) or are from different cells (FALSE).
?combine_cds
Wrapping Up
-Today we learned how to:
- create the data structures (CDS) that contain our single cell RNA-sequencing data starting with different data formats
- subset a CDS on a particular column value
- merge multiple CDSs
Next class we will talk more about how to analyze our data with Monocle.